
The Potential Role of Genic-SSRs in Driving Ecological Adaptation Diversity in Caragana Plants
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Sequencing Outputs and Assembly
2.2. Identification of Polymorphic Genic-SSRs
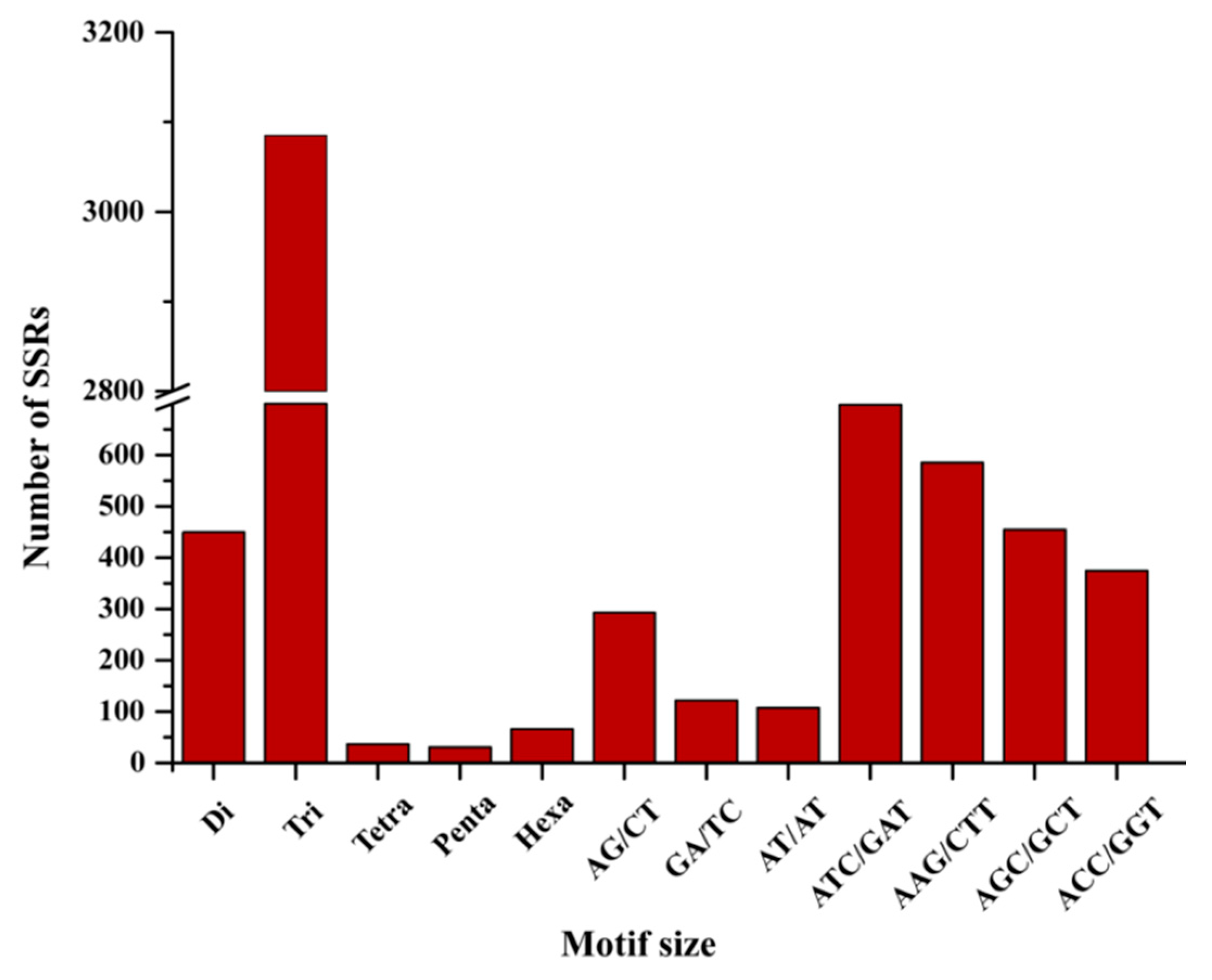
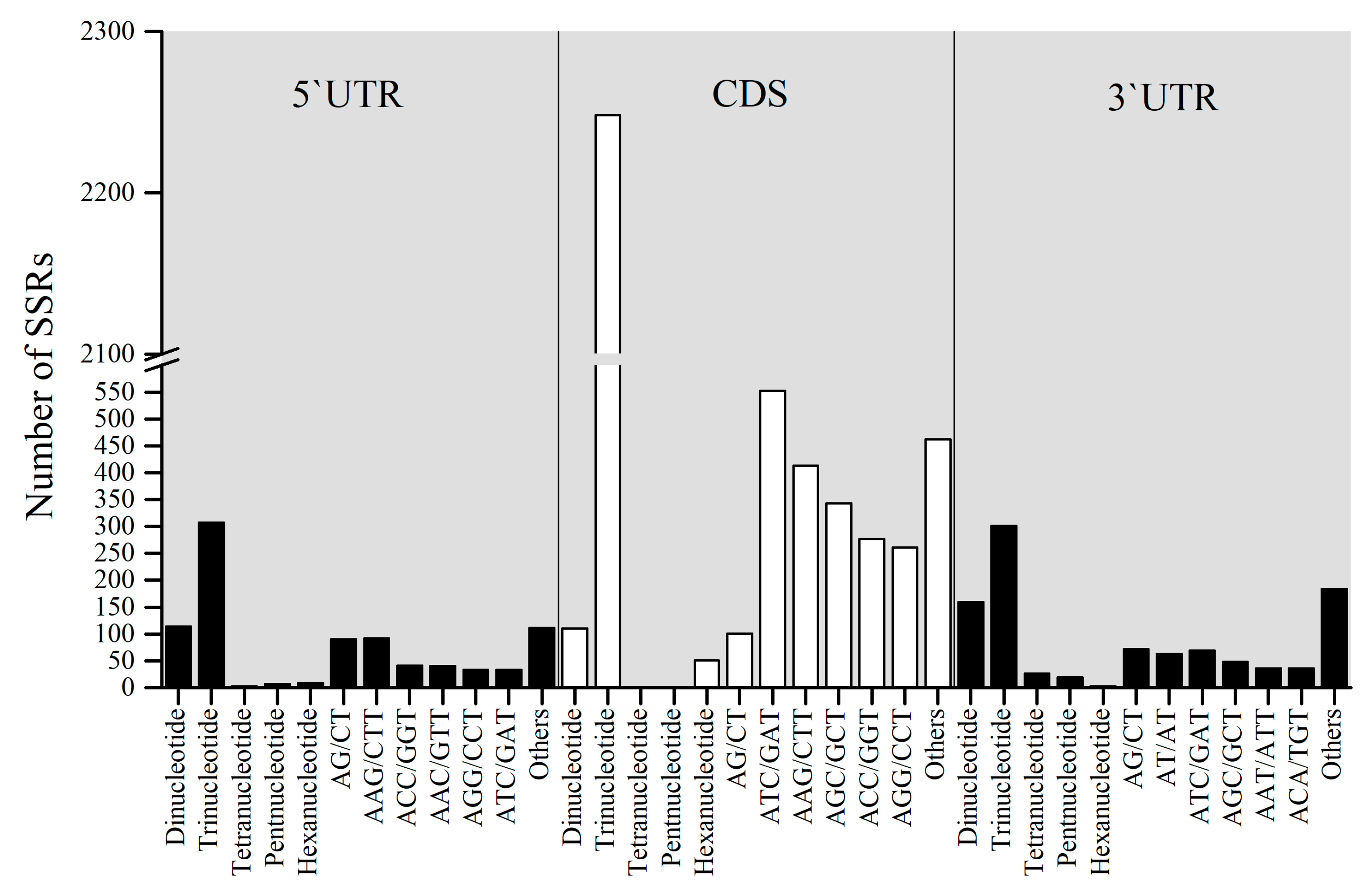
2.3. Location Prediction and Frequency Analysis of the Polymorphic Genic-SSRs
2.4. Functional Annotation of the Polymorphic SSR-Containing Sequences
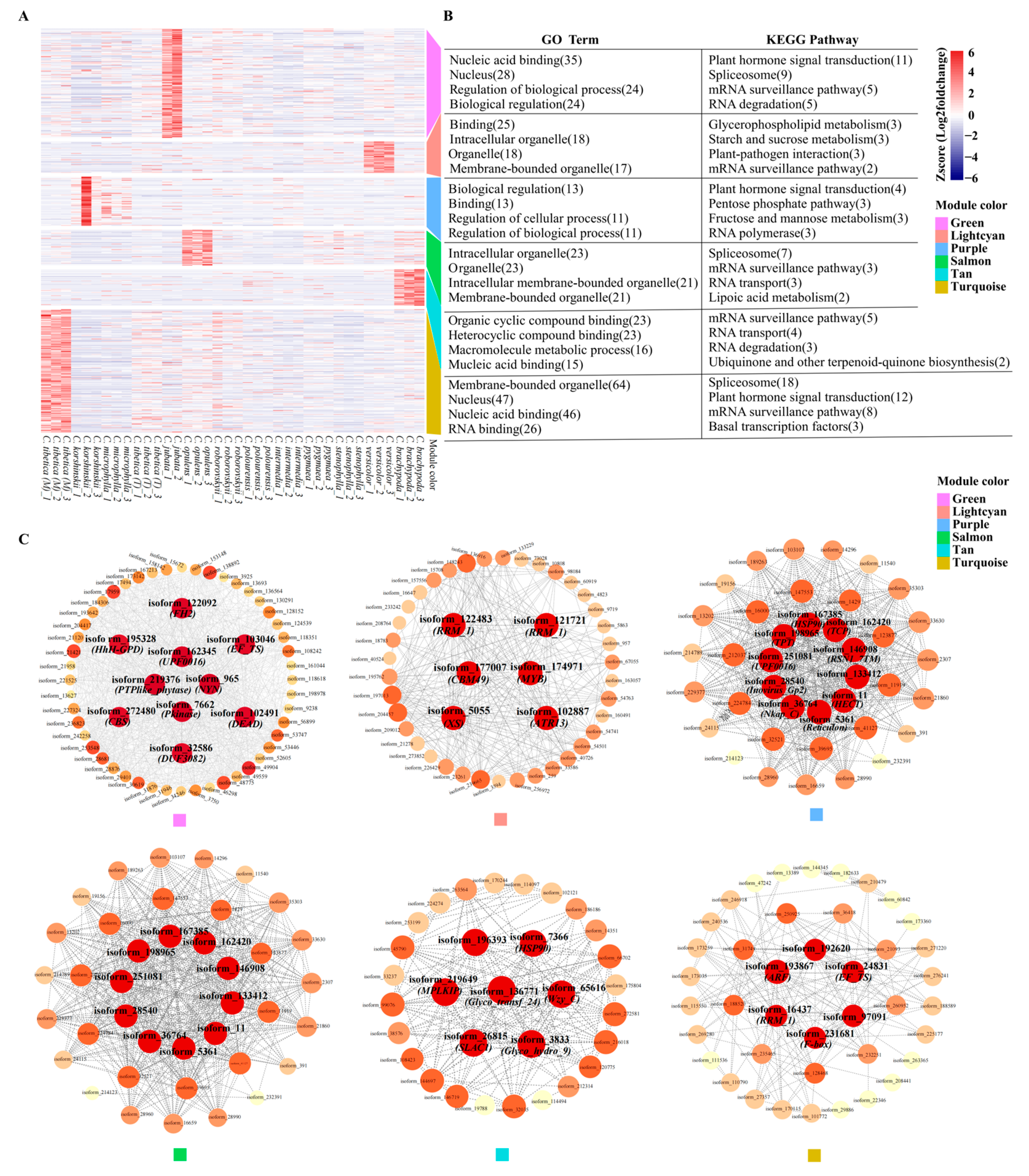
2.5. WGCNA Analysis of Polymorphic Genic-SSRs Related Differentially Expressed Genes on Functional Traits
2.6. WGCNA Analysis of Polymorphic Genic-SSRs Related Differentially Expressed Genes on Climate Factors
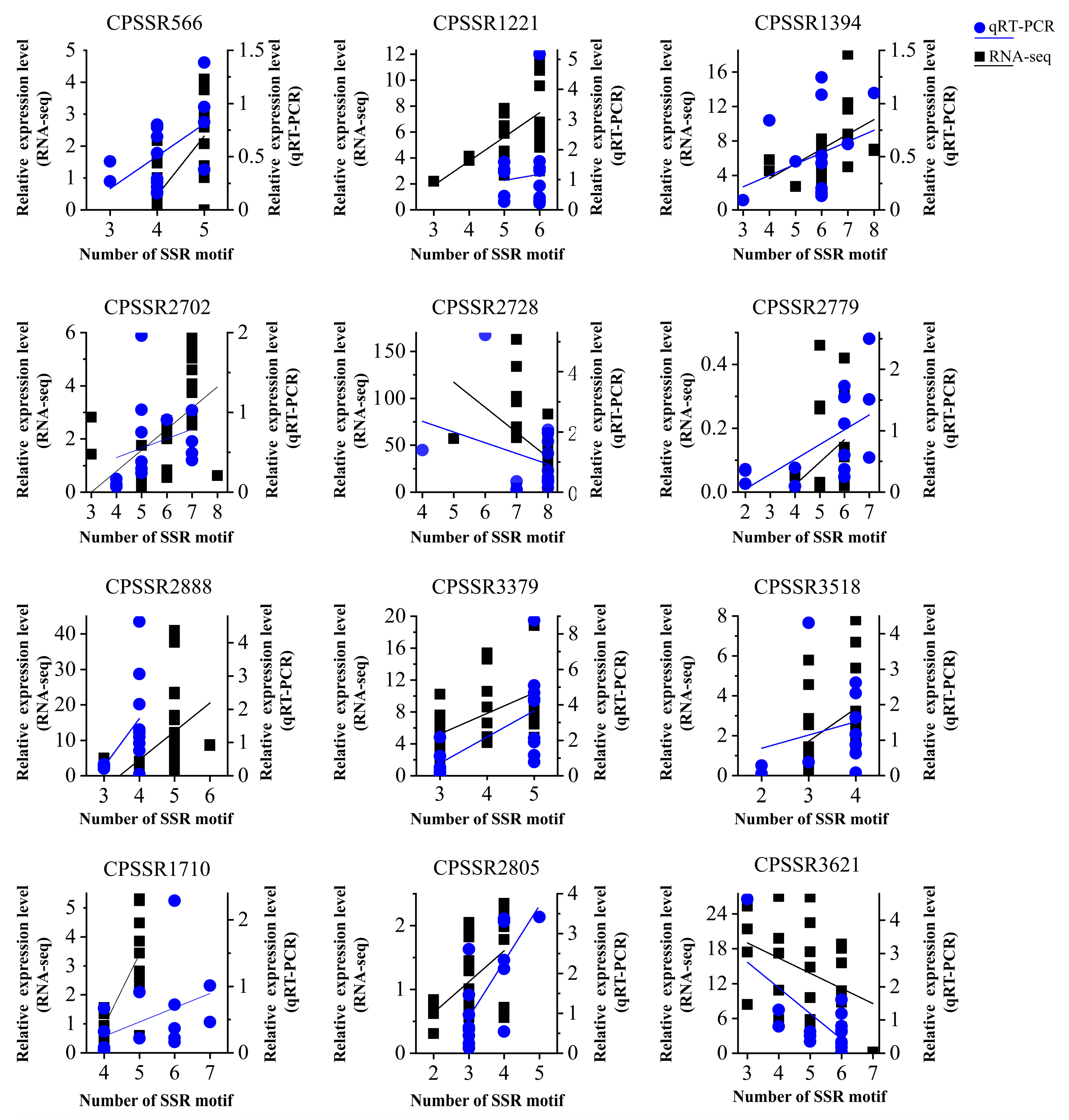
2.7. Correlation Analysis between SSR Polymorphism and Gene Expression
2.8. Experimental Validation of SSR Polymorphism and Their Effect on Gene Expression
3. Discussion
3.1. Polymorphic Genic-SSRs Provide the Genetic Basis for the Evolution of Ecological Adaptability of Caragana
3.2. Potential Roles of Polymorphic Genic-SSRs in the Formation of Ecological Adaptations in Caragana
4. Materials and Methods
4.1. Plant Materials
4.2. Full-Length Transcriptome Sequencing and Bioinformatics Analysis
4.3. Short-Reads Transcriptome Sequencing and Bioinformatics Analysis
4.4. Identification and Characteristic Analysis of Polymorphic Genic-SSRs
4.5. Weighted Gene Co-Expression Network Analysis and Hub Gene Identifying
4.6. Effect of Genic-SSRs’ Allele Length on Gene Expression
4.7. Validation of SSR Polymorphism and Their Effect on Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, S.; Wang, J.; Guo, Z.; He, Z.; Shi, S. Genomic convergence in the adaptation to extreme environments. Plant Commun. 2020, 1, 100117. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Q.; Asim, M.; Zhang, R.; Khan, R.; Farooq, S.; Wu, J. Transcription factors interact with ABA through gene expression and signaling pathways to mitigate drought and salinity stress. Biomolecules 2021, 11, 1159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of plant responses to salt stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef] [PubMed]
- Keil, L.; Mehlmer, N.; Cavelius, P.; Garbe, D.; Haack, M.; Ritz, M.; Awad, D.; Brück, T. The time-resolved salt stress response of Dunaliella tertiolecta—A comprehensive system biology perspective. Int. J. Mol. Sci. 2023, 24, 15374. [Google Scholar] [CrossRef] [PubMed]
- Wenne, R. Microsatellites as molecular markers with applications in exploitation and conservation of aquatic animal populations. Genes 2023, 14, 808. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.D.; Eggler, P.; Seaton, G.; Rossetto, M.; Ablett, E.M.; Lee, L.S.; Henry, R.J. Analysis of SSRs derived from grape ESTs. Theor. Appl. Genet. 2000, 100, 723–726. [Google Scholar] [CrossRef]
- Hamm, T.P.; Boggess, S.L.; Kandel, J.S.; Staton, M.E.; Huff, M.L.; Hadziabdic, D.; Shoemaker, D.; Adamczyk, J.J., Jr.; Nowicki, M.; Trigiano, R.N. Development and characterization of 20 genomic SSR markers for ornamental cultivars of Weigela. Plants 2022, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Ranathunge, C.; Wheeler, G.L.; Chimahusky, M.E.; Perkins, A.D.; Pramod, S.; Welch, M.E. Transcribed microsatellite allele lengths are often correlated with gene expression in natural sunflower populations. Mol. Ecol. 2020, 29, 1704–1716. [Google Scholar] [CrossRef]
- Parmar, R.; Seth, R.; Sharma, R.K. Genome-wide identification and characterization of functionally relevant microsatellite markers from transcription factor genes of Tea (Camellia sinensis (L.) O. Kuntze). Sci. Rep. 2022, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Streelman, J.T.; Kocher, T.D. Microsatellite variation associated with prolactin expression and growth of salt-challenged tilapia. Physiol. Genom. 2002, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Mao, M.; Wan, D.; Yang, Q.; Yang, F.; Mandlaa; Li, G.; Wang, R. Identification of the WRKY gene family and functional analysis of two genes in Caragana intermedia. BMC Plant Biol. 2018, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ni, Y.; Li, J.; Huang, L.; Li, H.; Chen, H.; Liu, C. Multiple configurations of the plastid and mitochondrial genomes of Caragana spinosa. Planta 2023, 258, 98. [Google Scholar] [CrossRef] [PubMed]
- Ramyar, R.; Ackerman, A.; Johnston, D.M. Adapting cities for climate change through urban green infrastructure planning. Cities 2021, 117, 103316. [Google Scholar] [CrossRef]
- Kisvarga, S.; Horotán, K.; Wani, M.A.; Orlóci, L. Plant Responses to Global Climate Change and Urbanization: Implications for Sustainable Urban Landscapes. Horticulturae 2023, 9, 1051. [Google Scholar] [CrossRef]
- Yao, G.Q.; Nie, Z.F.; Turner, N.C.; Li, F.M.; Gao, T.P.; Fang, X.W.; Scoffoni, C. Combined high leaf hydraulic safety and efficiency provides drought tolerance in Caragana species adapted to low mean annual precipitation. New Phytol. 2021, 229, 230–244. [Google Scholar] [CrossRef]
- Luo, Y.L.; Ma, W.H.; Zhang, X.Y.; Su, C.; Shi, Y.B.; Zhao, L.Q. Variation of functional traits of alternative distributed Caragana species along environmental gradients in Nei Mongol. Chin. J. Plant Ecol. 2022, 46, 1364–1375. [Google Scholar] [CrossRef]
- Khan, S.; Nazir, M.; Raiz, N.; Saleem, M.; Zengin, G.; Fazal, G.; Saleem, H.; Mukhtar, M.; Tousif, M.I.; Tareen, R.B.; et al. Phytochemical profiling, in vitro biological properties and in silico studies on Caragana ambigua stocks (Fabaceae): A comprehensive approach. Ind. Crops Prod. 2019, 131, 117–124. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, L.; Yang, G.; Gao, Y.; Li, H. Simulation and prediction of the geographical distribution of five Caragana species in the north temperate zone. Environ. Monit. Assess. 2023, 195, 1427. [Google Scholar] [CrossRef]
- Bi, M.H.; Jiang, C.; Yao, G.Q.; Turner, N.C.; Scoffoni, C.; Fang, X.-W. Rapid drought-recovery of gas exchange in Caragana species adapted to low mean annual precipitation. Plant Cell Environ. 2023, 46, 2296–2309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Xiang, X.G.; Xue, J.J.; Sanderson, S.C.; Fritsch, P.W. Himalayan uplift shaped biomes in Miocene temperate Asia: Evidence from leguminous Caragana. Sci. Rep. 2016, 6, 36528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Xue, J.J.; Zhang, Q.; Sanderson, S.C. Inferring ancestral distribution area and survival vegetation of Caragana (Fabaceae) in Tertiary. Plant Syst. Evol. 2015, 301, 1831–1842. [Google Scholar] [CrossRef]
- Li, J.; Gao, Y.B.; Wu, J.B.; Guo, H. Vessel features of three Caragana species in their natural habitats in the inner Mongolia Plateau of China. Nord. J. Bot. 2007, 25, 342–350. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yang, J.; Yang, M.B. Changes of osmotic adjustment solute content of the 5 species in Caragana genus. Chin. J. Acta Sci. Nat. Univ. NeiMongol 2005, 36, 677–682. (In Chinese) [Google Scholar]
- Duan, L.; Yang, X.; Liu, P.; Johnson, G.; Wen, J.; Chang, Z. A molecular phylogeny of Caraganeae (Leguminosae, Papilionoideae) reveals insights into new generic and infrageneric delimitations. PhytoKeys 2016, 70, 111–137. [Google Scholar] [CrossRef]
- Sharma, V.; Yuan, M.; Yin, X.; Gao, B.; Gu, R.; Jiang, G. The chloroplasts genomic analyses of four specific Caragana species. PLoS ONE 2022, 17, e0272990. [Google Scholar] [CrossRef]
- Duan, L.; Li, S.-J.; Su, C.; Sirichamorn, Y.; Han, L.-N.; Ye, W.; Lôc, P.K.; Wen, J.; Compton, J.A.; Schrire, B.; et al. Phylogenomic framework of the IRLC legumes (Leguminosae subfamily Papilionoideae) and intercontinental biogeography of tribe Wisterieae. Mol. Phylogenet. Evol. 2021, 163, 107235. [Google Scholar] [CrossRef]
- Guo, Q.; Shi, Y.J.; Wei, Z.W.; Yang, Z.H. Genetic diversity analysis by SSR marker of fourteen species of Caragana Fabr. in He-Xi corridor area of Gansu. Acta Agrestia Sin. 2008, 3, 23–29. (In Chinese) [Google Scholar]
- Huang, W.D.; Zhao, X.Y.; Zhao, X.; Li, Y.; Lian, J. Effects of environmental factors on genetic diversity of Caragana microphylla in Horqin Sandy Land, northeast China. Ecol. Evol. 2016, 6, 8256–8266. [Google Scholar] [CrossRef]
- Xu, B.; Sun, G.; Wang, X.; Lu, J.; Wang, I.J.; Wang, Z. Population genetic structure is shaped by historical, geographic, and environmental factors in the leguminous shrub Caragana microphylla on the Inner Mongolia Plateau of China. BMC Plant Biol. 2017, 17, 200. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Yang, J.; Yang, M.B.; Qing, H. Genetic diversity of Caragana species of the Ordos Plateau in China. Plant Syst. Evol. 2012, 298, 801–809. [Google Scholar] [CrossRef]
- Long, Y.; Wang, Y.; Wu, S.; Wang, J.; Tian, X.; Pei, X. De novo assembly of transcriptome sequencing in Caragana korshinskii Kom. and characterization of EST-SSR markers. PLoS ONE 2015, 10, e0115805. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, C.; Li, Y.; Zhang, J.; Sun, J.; Chen, Y.; Tian, C.; Su, X.; Lu, M.; Liang, C.; et al. Effects of drought and salt-stresses on gene expression in Caragana korshinskii seedlings revealed by RNA-seq. BMC Genom. 2016, 17, 200. [Google Scholar] [CrossRef]
- Zhu, J.; Li, W.; Yang, W.; Qi, L.; Han, S. Identification of microRNAs in Caragana intermedia by high-throughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013, 32, 1339–1349. [Google Scholar] [CrossRef]
- Ning, P.; Wang, J.; Zhou, Y.; Gao, L.; Wang, J.; Gong, C. Adaptional evolution of trichome in Caragana korshinskii to natural drought stress on the Loess Plateau, China. Ecol. Evol. 2016, 6, 3786–3795. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Liu, X.; Ding, H.; Dai, Z.; Niu, X.; Zhao, L. Hormonal balance, photosynthesis, and redox reactions in the leaves of Caragana korshinskii Kom. under water deficit. Plants 2023, 12, 2076. [Google Scholar] [CrossRef]
- Zhao, M.; Shu, G.; Hu, Y.; Cao, G.; Wang, Y. Pattern and variation in simple sequence repeat (SSR) at different genomic regions and its implications to maize evolution and breeding. BMC Genom. 2023, 24, 136. [Google Scholar] [CrossRef]
- Sawaya, S.; Bagshaw, A.; Buschiazzo, E.; Kumar, P.; Chowdhury, S.; Black, M.A.; Gemmell, N. Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS ONE 2013, 8, e54710. [Google Scholar] [CrossRef]
- Lopez, L.; Barreiro, R.; Fischer, M.; Koch, M.A. Mining microsatellite markers from public expressed sequence tags databases for the study of threatened plants. BMC Genom. 2015, 16, 781. [Google Scholar] [CrossRef]
- Li, S.X.; Yin, T.M.; Wang, M.X.; Tuskan, G.A. Characterization of microsatellites in the coding regions of the Populus genome. Mol. Breed. 2010, 27, 59–66. [Google Scholar] [CrossRef]
- Biswas, M.K.; Bagchi, M.; Nath, U.K.; Biswas, D.; Natarajan, S.; Jesse, D.M.I.; Park, J.I.; Nou, I.S. Transcriptome wide SSR discovery cross-taxa transferability and development of marker database for studying genetic diversity population structure of Lilium species. Sci. Rep. 2020, 10, 18621. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.J.; Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [PubMed]
- de Santana Lopes, A.; Gomes Pacheco, T.; Nascimento da Silva, O.; do Nascimento Vieira, L.; Guerra, M.P.; Pacca Luna Mattar, E.; de Baura, V.A.; Balsanelli, E.; Maltempi de Souza, E.; de Oliveira Pedrosa, F.; et al. Plastid genome evolution in Amazonian açaí palm (Euterpe oleracea Mart.) and Atlantic forest açaí palm (Euterpe edulis Mart.). Plant Mol. Biol. 2021, 105, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Liu, S.; He, G.; Xie, G.; Gong, Y.; Zhu, N.; Xiao, C. De novo assembly of Iron-Heart Cunninghamia lanceolata transcriptome and EST-SSR marker development for genetic diversity analysis. PLoS ONE 2023, 18, e0293245. [Google Scholar] [CrossRef]
- Tan, L.Q.; Wang, L.Y.; Wei, K.; Zhang, C.C.; Wu, L.Y.; Qi, G.N.; Cheng, H.; Zhang, Q.; Cui, Q.M.; Liang, J.B. Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS ONE 2013, 8, e81611. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bhatia, S. A polymorphic (GA/CT)n- SSR influences promoter activity of Tryptophan decarboxylase gene in Catharanthus roseus L. Don. Sci. Rep. 2016, 6, 33280. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fang, Z.; Peng, H.; Zhou, J.; Liu, P.; Wang, Y.; Zhu, W.; Li, L.; Zhang, Q.; Chen, L.; et al. Application of high-throughput amplicon sequencing-based SSR genotyping in genetic background screening. BMC Genom. 2019, 20, 444. [Google Scholar] [CrossRef]
- Lu, Q. The capacity to form H-DNA cannot substitute for GAGA factor binding to a (CT)nmiddle dot(GA)n regulatory site. Nucleic Acids Res. 2003, 31, 2483–2494. [Google Scholar] [CrossRef]
- Punt, P.J.; Dingemanse, M.A.; Kuyvenhoven, A.; Soede, R.D.; Pouwels, P.H.; van den Hondel, C.A. Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 1990, 93, 101–109. [Google Scholar] [CrossRef]
- Sandaltzopoulos, R.; Mitchelmore, C.; Bonte, E.; Wall, G.; Becker, P.B. Dual regulation of the Drosophila hsp26 promoter in vitro. Nucleic Acids Res. 1995, 23, 2479–2487. [Google Scholar] [CrossRef]
- Panda, S.; Swain, S.K.; Sahu, B.P.; Sarangi, R. Insights into genome plasticity and gene regulation in Orientia tsutsugamushi through genome-wide mining of microsatellite markers. 3 Biotech 2023, 13, 366. [Google Scholar] [CrossRef]
- Gemayel, R.; Chavali, S.; Pougach, K.; Legendre, M.; Zhu, B.; Boeynaems, S.; van der Zande, E.; Gevaert, K.; Rousseau, F.; Schymkowitz, J.; et al. Variable glutamine-rich repeats modulate transcription factor activity. Mol. Cell 2015, 59, 615–627. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, X.; Kettenbach, A.N.; Mitchell, H.D.; Markillie, L.M.; Loros, J.J.; Dunlap, J.C. A crucial role for dynamic expression of components encoding the negative arm of the circadian clock. Nat. Commun. 2023, 14, 3371. [Google Scholar] [CrossRef]
- Golubov, A.; Yao, Y.; Maheshwari, P.; Bilichak, A.; Boyko, A.; Belzile, F.; Kovalchuk, I. Microsatellite instability in Arabidopsis increases with plant development. Plant Physiol. 2010, 154, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Ravigné, V.; Rodrigues, L.R.; Charlery de la Masselière, M.; Facon, B.; Kuczyński, L.; Radwan, J.; Skoracka, A.M.; Magalhães, S. Understanding the joint evolution of dispersal and host specialisation using phytophagous arthropods as a model group. Biol. Rev. Camb. Philos. Soc. 2024, 99, 219–237. [Google Scholar] [CrossRef]
- Vázquez, D.P.; Gianoli, E.; Morris, W.F.; Bozinovic, F. Ecological and evolutionary impacts of changing climatic variability. Biol. Rev. Camb. Philos. Soc. 2017, 92, 22–42. [Google Scholar] [CrossRef]
- Carvajal, D.E.; Loayza, A.P.; Rios, R.S.; Gianoli, E.; Squeo, F.A. Population variation in drought-resistance strategies in a desert shrub along an aridity gradient: Interplay between phenotypic plasticity and ecotypic differentiation. Perspect. Plant Ecol. Evol. Syst. 2017, 29, 12–19. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Z.; Li, H.; Yang, J.; Liu, B.; Yang, Y. Contrasting patterns of genetic and phenotypic divergence of two sympatric congeners, Phragmites australis and P. hirsuta, in heterogeneous habitats. Front. Plant Sci. 2023, 14, 1299128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, N.; Zhang, F.; Wu, T.; He, X.; Wang, J.; Li, Y. Intraspecific variations of leaf hydraulic, economic, and anatomical traits in Cinnamomum camphora along an urban-rural gradient. Sci. Total Environ. 2023, 904, 166741. [Google Scholar] [CrossRef]
- Lázaro-Nogal, A.; Matesanz, S.; Godoy, A.; Pérez-Trautman, F.; Gianoli, E.; Valladares, F.; Cornwell, W. Environmental heterogeneity leads to higher plasticity in dry-edge populations of a semi-arid Chilean shrub: Insights into climate change responses. J. Ecol. 2015, 103, 338–350. [Google Scholar] [CrossRef]
- Matesanz, S.; Gianoli, E.; Valladares, F. Global change and the evolution of phenotypic plasticity in plants. Ann. N. Y. Acad. Sci. 2010, 1206, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Wei, Y.M.; Yan, Z.H.; Zheng, Y.L. EST-SSR DNA polymorphism in durum wheat (Triticum durum L.) collections. J. Appl. Genet. 2007, 48, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Qiu, T.; Li, L.; Cui, S. De novo full-length transcriptome analysis of two ecotypes of Phragmites australis (swamp reed and dune reed) provides new insights into the transcriptomic complexity of dune reed and its long-term adaptation to desert environments. BMC Genom. 2023, 24, 180. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Zhang, S.; Liu, J.Y.; Zhao, C.; Wu, J.; Cao, Y.; Fu, C.; Han, X.; He, H.; Zhao, Q. MYB20, MYB42, MYB43, and MYB85 regulate phenylalanine and lignin biosynthesis during secondary cell wall formation. Plant Physiol. 2020, 182, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Dalton, S.; Roberts, L.A.; Moron-Garcia, O.M.; Iacono, R.; Kosik, O.; Gallagher, J.A.; Bosch, M. Modified expression of ZmMYB167 in Brachypodium distachyon and Zea mays leads to increased cell wall lignin and phenolic content. Sci. Rep. 2019, 9, 8800. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.Y.; Wang, X.Q.; Li, C.F.; Lu, W.; Yang, L.; Jiang, Y.; Luo, K. Functional characterization of the poplar R2R3-MYB transcription factor PtoMYB216 involved in the regulation of lignin biosynthesis during wood formation. PLoS ONE 2013, 8, e76369. [Google Scholar] [CrossRef]
- Davière, J.M.; Wild, M.; Regnault, T.; Baumberger, N.; Eisler, H.; Genschik, P.; Achard, P. Class I TCP-DELLA interactions in inflorescence shoot apex determine plant height. Curr. Biol. 2014, 24, 1923–1928. [Google Scholar] [CrossRef]
- Challa, K.R.; Aggarwal, P.; Nath, U. Activation of YUCCA5 by the transcription factor TCP4 integrates developmental and environmental signals to promote hypocotyl elongation in Arabidopsis. Plant Cell 2016, 28, 2117–2130. [Google Scholar] [CrossRef]
- Meng, X.W.; Yang, T.; Liu, J.; Zhao, M.; Wang, J. Genome-wide identification and evolution of HECT genes in wheat. PeerJ 2020, 8, e10457. [Google Scholar] [CrossRef]
- Shen, C.J.; Wang, S.K.; Bai, Y.H.; Wu, Y.; Zhang, S.; Chen, M.; Guilfoyle, T.J.; Wu, P.; Qi, Y. Functional analysis of the structural domain of ARF proteins in rice (Oryza sativa L.). J. Exp. Bot. 2010, 61, 3971–3981. [Google Scholar] [CrossRef]
- Attia, K.A.; Abdelkhalik, A.F.; Ammar, M.H.; Wei, C.; Yang, J.; Lightfoot, D.A.; El-Sayed, W.M.; El-Shemy, H.A. Antisense phenotypes reveal a functional expression of OsARF1, an auxin response factor, in transgenic rice. Curr. Issues Mol. Biol. 2009, 11 (Suppl. S1), i29–i34. [Google Scholar] [PubMed]
- Zhang, S.; Wang, S.; Xu, Y.; Yu, C.; Shen, C.; Qian, Q.; Geisler, M.; Jiang, D.A.; Qi, Y. The auxin response factor, OsARF19, controls rice leaf angles through positively regulating OsGH3-5 and OsBRI1. Plant Cell Env. 2015, 38, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Hu, L.; Punta, M.; Bruni, R.; Hillerich, B.; Kloss, B.; Rost, B.; Love, J.; Siegelbaum, S.A.; Hendrickson, W.A. Homologue structure of the SLAC1 anion channel for closing stomata in leaves. Nature 2010, 467, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeong, H.S.; Jung, S.; Hwang, J.; Le, C.T.H.; Jun, S.-H.; Du, E.J.; Kang, K.; Kim, B.-G.; Lim, H.-H.; et al. Cryo-EM structures of the plant anion channel SLAC1 from Arabidopsis thaliana suggest a combined activation model. Nat. Commun. 2023, 14, 7345. [Google Scholar] [CrossRef]
- Hedrich, R.; Geiger, D. Biology of SLAC1-type anion channels—From nutrient uptake to stomatal closure. New Phytol. 2017, 216, 46–61. [Google Scholar] [CrossRef]
- Baute, J.; Polyn, S.; De Block, J.; Blomme, J.; Van Lijsebettens, M.; Inze, D. F-box protein FBX92 affects leaf size in Arabidopsis thaliana. Plant Cell Physiol 2017, 58, 962–975. [Google Scholar] [CrossRef]
- Virdi, K.S.; Sreekanta, S.; Dobbels, A.; Haaning, A.; Jarquin, D.; Stupar, R.M.; Lorenz, A.J.; Muehlbauer, G.J. Branch angle and leaflet shape are associated with canopy coverage in soybean. Plant Genome 2023, 16, e20304. [Google Scholar] [CrossRef]
- Tomar, S.; Subba, A.; Bala, M.; Singh, A.K.; Pareek, A.; Singla-Pareek, S.L. Genetic conservation of CBS domain containing protein family in Oryza species and their association with abiotic stress responses. Int. J. Mol. Sci. 2022, 23, 1687. [Google Scholar] [CrossRef]
- Guo, F.Y.; Chang, J.; Lu, Q.W. Genome-wide analysis of CBS gene family in Hexpaploid Wheat. Chin. J. Triticeae Crops 2020, 40, 425–433. (In Chinese) [Google Scholar]
- Johnson, K.L.; Kibble, N.A.; Bacic, A.; Schultz, C.J. A fasciclin-like arabinogalactan-protein (FLA) mutant of Arabidopsis thaliana, fla1, shows defects in shoot regeneration. PLoS ONE 2011, 6, e25154. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, C.P.; Mansfield, S.D.; Stachurski, Z.H.; Evans, R.; Southerton, S.G. Fasciclin-like arabinogalactan proteins: Specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J. 2010, 62, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Attia, K.; Wei, C.; Li, K.; He, G.; Su, W.; Zhang, Q.; Qian, X.; Yang, J. Overexpression of the rFCA RNA recognition motif affects morphologies modifications in rice (Oryza sativa L.). Biosci. Rep. 2007, 27, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Xia, E.H.; Yao, Q.Y.; Zhang, H.B.; Jiang, J.J.; Zhang, L.P.; Gao, L.Z. CandiSSR: An efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 2015, 6, 1171. [Google Scholar] [CrossRef]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Vinces, M.D.; Legendre, M.; Caldara, M.; Hagihara, M.; Verstrepen, K.J. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 2009, 324, 1213–1216. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Longitude (E) | Latitude (N) | Altitude (m) |
|---|---|---|---|
| C. tibetica (Inner Mongolia) | 108°19′09.63″ | 41°48′00.61″ | 1411 |
| C. korshinskii | 119°50′38.87″ | 45°14′05.92″ | 1149 |
| C. microphylla | 115°42′07.48″ | 43°31′48.47″ | 1191 |
| C. tibetica (Tibet) | 87°14′02.1″ | 28°36′15.9″ | 4231 |
| C. jubata | 105°56′53.12″ | 38°50′09.92″ | 3472 |
| C. opulens | 112°00′35.78″ | 40°25′27.07″ | 1906 |
| C. roborovskyii | 105°48′00.58″ | 38°40′16.90″ | 2004 |
| C. polourensis | 75°04′05.9″ | 39°42′22.1″ | 2130 |
| C. intermedia | 112°58′10.87″ | 43°11′22.24″ | 1058 |
| C. pygmaea | 112°25′54.79″ | 42°34′1.61″ | 1213 |
| C. stenophylla | 110°10′16.62″ | 41°40′41.93″ | 1573 |
| C. versicolor | 85°32′48.6″ | 28°45′35.1″ | 4625 |
| C. brachypoda | 107°54′59.61″ | 42°02′46.33″ | 1310 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Chen, X.; Meng, Y.; Niu, M.; Jia, Y.; Huang, L.; Ma, W.; Liang, C.; Li, Z.; Zhao, L.; et al. The Potential Role of Genic-SSRs in Driving Ecological Adaptation Diversity in Caragana Plants. Int. J. Mol. Sci. 2024, 25, 2084. https://doi.org/10.3390/ijms25042084
Wang Q, Chen X, Meng Y, Niu M, Jia Y, Huang L, Ma W, Liang C, Li Z, Zhao L, et al. The Potential Role of Genic-SSRs in Driving Ecological Adaptation Diversity in Caragana Plants. International Journal of Molecular Sciences. 2024; 25(4):2084. https://doi.org/10.3390/ijms25042084
Chicago/Turabian StyleWang, Qinglang, Xing’er Chen, Yue Meng, Miaomiao Niu, Yuanyuan Jia, Lei Huang, Wenhong Ma, Cunzhu Liang, Zhiyong Li, Liqing Zhao, and et al. 2024. "The Potential Role of Genic-SSRs in Driving Ecological Adaptation Diversity in Caragana Plants" International Journal of Molecular Sciences 25, no. 4: 2084. https://doi.org/10.3390/ijms25042084